
David G. DeNardo, PhD
Professor; Section Co-Director - Molecular Oncology
- Phone: 314-362-9524
- Fax: 314-747-2797
- Email: ddenardo@nospam.wustl.edu
Address:
Division of Oncology
MSC 8069-0004-07
Washington University
660 South Euclid Avenue
St. Louis, MO 63110
Room 7611 BJCIH (office)
Room 7207 BJCIH (lab)
Ph: 314-362-9534 (lab)
- Immune response
- Metastasis
- Breast cancer
- Pancreatic cancer
In general, my laboratory looks at understanding how immune responses shape and are shaped by malignancies. Where possible, our hope is to exploit these weaknesses to improve patient outcomes by enhancing response to conventional, targeted or immunotherapeutics. Currently, the laboratory focuses primarily on pancreas and breast cancers.
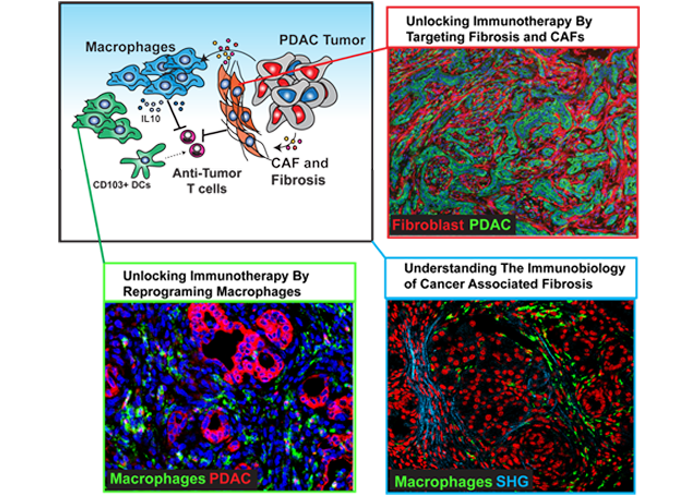
Example Project: The prognosis for pancreatic cancer (PDAC) patients is dismal. The application of immunotherapy holds the promise to revolutionize PDAC patient outcomes. Unfortunately, attempts at immunotherapy in PaC, to date, have not achieved significant clinical benefits as single agents. This is likely due to the presence of a uniquely suppressive tumor microenvironment (TME) that is dominant in most pancreatic ductal PDAC. Two major drivers of this tumor protective microenvironment include a dense fibrotic tumor stroma and robust infiltration by tumor-supportive myeloid cells. High stromal density provides a barrier to T cell infiltration and function. These data suggest that we could improve PaC patient outcomes, if we could identify therapeutics that reprogram the protective TME to facilitate immunotherapy. One such approach is to target the oncogenic pathways that induce the fibrotic and suppressive PDAC TME.
Recently, we have identified focal adhesion kinase (FAK)-1, which is hyper-activated as a major driver of the fibrotic and inflammatory (fibro-inflammatory) TME of PDAC tumors. We have discovered that in human PDAC, elevated FAK activity correlates with higher fibrosis levels, excessive myeloid cell infiltration, and poor T cell responses. These data suggest that FAK is a key mediator of the fibro-inflammatory microenvironment that blocks immunotherapeutic efficacy. FAK inhibition dramatically reduced fibrosis and inflammatory myeloid cell infiltration, and improved T cell function in PaC mouse models.
Key papers or links:
Jiang et al. Nature Medicine. (2016). PMID: 27376576
Jiang et al. Cancer Immunol Immunother. (2017). PMID: 28451791
Seton-Rogers S. Nature Reviews Cancer. 16, 480–481 (2016)
Clinical Trial NCT02546531
2) Understanding the differential effects of Macrophage origin on tumor fibrosis, immunity and progression
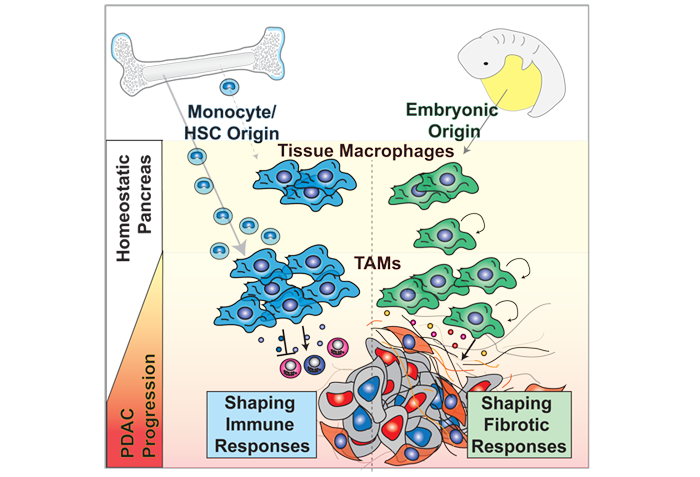
Example Project: Tumor-associated macrophages (TAMs) are essential components of the cancer microenvironment and play critical roles in the regulation of tumor progression. Optimal therapeutic intervention requires in-depth understanding of the sources that sustain macrophages in malignant tissues. In this study, we systemically investigated the ontogeny of TAMs in murine pancreatic ductal adenocarcinoma (PDAC) models. We identified both inflammatory monocytes and tissue-resident macrophages as sources of TAMs. Unexpectedly, significant portions of pancreas resident macrophages originate during embryonic development and expand through in situ proliferation during tumor progression. While monocyte-derived TAMs play more potent roles in antigen presentation, embryonically derived TAMs exhibit a pro-fibrotic transcriptional profile suggesting their role in producing and remodeling extracellular matrix molecules. Collectively, these findings uncovered the heterogeneity of TAM origin and functions, and could provide therapeutic insight for PDAC treatment.
Key papers or links:
Zhu et al. Immunity (in press 2017)
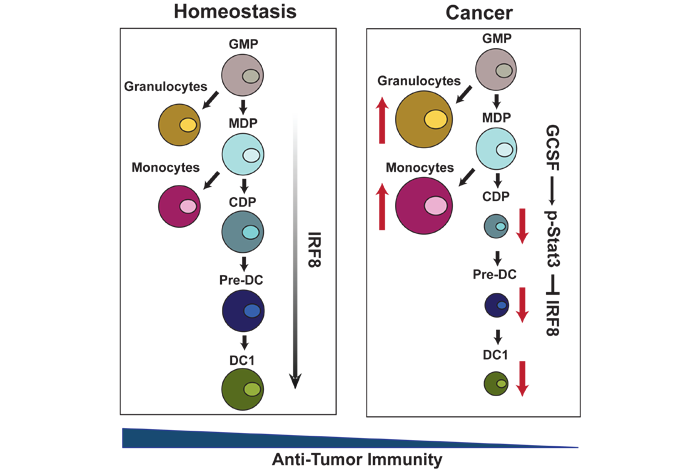
Example Project: Tumors must evade immune-surveillance in order to progress. To this end, tumor-induced inflammation expands immune-suppressive immature granulocytes and monocytes, while limiting the functions of immune-stimulatory conventional dendritic cells (cDCs). cDC1s, a cDC subset, activate CD8+ T cells through antigen cross presentation and are marked by CD141 in humans and CD103 or CD8α in mice. cDC1s support anti-tumor CD8+ T cell responses, but cDC1 numbers are limited within the tumor environment. To understand how cDC1s are restricted in the tumor, we interrogated their development in the bone marrow (BM). We found the program responsible for the expansion of immature granulocytes and monocytes from progenitors in the BM is simultaneously suppressing the generation of cDC1s, decreasing the systemic numbers of cDC1s in breast and pancreatic cancer patients. We found that tumor-induced granulocyte-stimulating factor (GCSF) mediates the down-regulation of the transcription factor IFN regulatory factor 8 (IRF8) in BM progenitors. IRF8 loss reprograms the BM progenitors, reducing their potential to develop into cDC1s. We demonstration, independent of monocyte and granulocyte expansion, that tumor-induced loss of cDC1s reduces the systemic ability to mount anti-tumor CD8+ T cell responses. Further, systemic decreases in cDC1s in breast cancer patients correlate with reduced response to chemotherapy. These data suggest a new mechanism of immune-escape whereby tumors down-regulate cDC1 differentiation from BM progenitors to impair anti-tumor CD8+ T cell immunity.
Key papers or links:
Coming soon
Project 1 of the Washington University Pancreas SPORE:
Immunotherapy against PDAC has struggled to achieve significant clinical benefit as single agents in PDAC. This is likely due to the presence of an immunosuppressive tumor microenvironment. Critical drivers of this immunosuppressive microenvironment are tumor-infiltrating inflammatory monocytes (IMs) and macrophages (TAMs). Thus, high numbers of these cells correlate with early metastatic relapse and poor survival in pancreatic cancer. Therefore, approaches that reprogram myeloid responses to potentiate protective antitumor immunity hold significant therapeutic potential.
We and other groups have demonstrated that mobilization and tumor infiltration of IMs and TAMs can promote local immunosuppression, and resistance to cytotoxic therapy. Signaling through C-C chemokine receptor type 2 (CCR2) is critical for the mobilization of IMs and their recruitment to inflamed tissues. Our recently published reports clearly illustrate that blockade of IM recruitment using a novel CCR2 inhibitor, PF-04136309 (CCR2i), slows tumor progression, improves responses to chemotherapy and prevents metastasis in mouse models of PDAC. Based on these exciting and provocative data, we initiated a Phase Ib/II clinical trial targeting the CCR2 signaling pathway in patients with locally advanced PDAC. In this trial, we have observed a remarkable 48.5% response rate in the 33 patients treated with CCR2i + FOLFIRINOX. Additionally, this regimen was well tolerated (safe). These responses appear to be correlated with a marked reduction in circulating CCR2+ IMs as well as decreased immune suppressive gene expression profiles in the primary tumor microenvironment. Paralleling these clinical data, our published pre-clinical studies found that CCR2 blockade overcomes immune suppression to reinitiate anti-tumor responses via CD8+ CTLs. Intriguingly, we’ve discovered that CCR2 blockade in both human patients and mouse models leads to the up-regulation of T cell checkpoint pathways, including programmed cell death-1 (PD1) and its ligands. These data suggest that we might find unique therapeutic synergy between CCR2 inhibition and PD1-based immunotherapies.
Biosketch
Education
- 2010-2005: Postdoctoral Scholar, Department of Pathology, University of California, San Francisco, CA
- 2005-2000: PhD in Cell Biology, Baylor College of Medicine, Houston, TX
- 1999-1995: BS in Biology, Willamette University, Salem, OR
Academic Positions & Employment
- present-2021: Professor, Departments of Medicine and Pathology & Immunology, Washington University, St. Louis, MO
- 2021-2017: Associate Professor, Departments of Medicine and Pathology & Immunology, Washington University, St. Louis, MO
- 2016-2011: Assistant Professor, Departments of Medicine and Pathology & Immunology, Washington University, St. Louis, MO
- 2001-2000: Research Associate (Genomics), InCyte Genomics, Palo Alto, CA
- 1992-1990: Research Assistant (Part-Time), University of California, Davis, CA
University Committees
- present-2021: IAB member, WUSM Leukemia SPORE
- present-2020: IAB member, WUSM Endo Cancer pre-SPORE
- present-2019: Co-Director, WUSM Pancreas Cancer SPORE
- present-2019: Molecular Genetics Graduate Program, Member
- present-2018: Immunology Graduate Program, Member
- present-2017: SCC – Basic Science Research Working Group
- present-2017: ACS-IRG Grant Review Committee
- present-2016: Administrative Co-leader, WUSM Pancreas Cancer SPORE
- present-2015: CHIIPs, Member
- present-2015: C4 Program, Siteman Cancer Center, Member
- present-2014: Musculoskeletal Research Center, Member
- present-2014: The Digestive Diseases Research Core Center, Member
- present-2014: ICCE Institute, Member
- present-2011: Molecular Cellular Biology Graduate Program, Member
- present-2011: Division of Biological and Biomedical Sciences, Member
- present-2011: Tumor Immunology Program, Siteman Cancer Center, Member
- 2021: Adhoc Research Integrity Committee
- 2018: Department of Medicine, Basic Research Working Group
Honors & Awards
- 2014: ACS Research Scholar Award (declined due to overlap with NIH-R01)
- 2014: AACR-PANCAN Research Scholar Award
- 2013: Cancer Research Foundation Young Investigator Award
- 2011: V Foundation V Scholar Award
- 2011: Edward Mallinckrodt Jr. Foundation Award
- 2010: Keystone Symposia Presenter Scholarship
- 2008: AACR-Aflac, Incorporated Scholar-in-Training Award
- 2008: Midwinter Conference of Immunologists, Ray Owen Young Investigator Award
- 2007: Ruth L. Kirschstein National Research Service Award, F32 Fellowship (declined)
- 2007: American Cancer Society/New England Division, Postdoctoral Fellowship Award
National Committees
- present-2021: EAB member, UNMC PDAC Cancer SPORE
- present-2018: NCI’s Pancreatic Cancer Microenvironment Network, Member
- present-2016: PANCAN Immunotherapy Working Group, Member
- 2020-2019: AACR National Meeting, Education Program Committee
- 2020-2019: AACR National Meeting, Scientific Program Subcommittee
- 2019: Chair, PANCAN Translational Research Program Grant Review
- 2018: NIH/NCI Special Emphasis Study Section, Adhoc Reviewer
- 2018: PANCAN Translational Research Program, Reviewer
- 2018: SITC, Immunotherapy Working Group, Member
- 2015: AACR National Meeting, Scientific Program Committee
- 2014: Worldwide Cancer Research Fund, Adhoc Reviewer
- 2014: Pancreatic Cancer Research Fund, Adhoc Reviewer
- 2014: DOD Breast Cancer Research Program, Reviewer
- 2013: American Institute for Cancer Research, Adhoc Reviewer
- 2013: NIH/NCI CII Study Section, Adhoc Reviewer
- 2012: DOD Breast Cancer Research Program, Reviewer
- 2011: AACR National Meeting, Scientific Program Committee