
Alessandro Vindigni, PhD
Professor; Section Co-Director - Molecular Oncology
- Phone: 314-273-8612
- Fax: 314-362-7086
- Email: avindigni@nospam.wustl.edu
Address:
Division of Oncology
Mail Stop 8125-0020-08
Washington University
660 South Euclid Avenue
St. Louis, MO 63110
Rooms 8844/8848 Clinical Sciences Research Building (lab)
Ph: 314-362-9221 (lab)
- DNA replication and repair
- Genome stability
- Cancer etiology
- DNA damaging chemotherapy
We focus on DNA replication and repair, and the roles of these pathways on cancer initiation, progression and response to chemotherapy and immunotherapy. We use a unique combination of biochemical, cellular, and electron microscopy approaches to study replication perturbations at single molecule resolutions. Combining these technologies, we have identified key pathways by which cancer cells respond to DNA damaging chemotherapy and have provided new insights on how to target these pathways to increase chemotherapeutic sensitivity.
We also are part of the newly established Siteman Center for Genome Integrity that fosters interactions between clinical and basic science investigators interested in genome integrity and DNA repair.
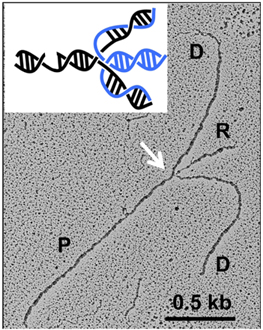
Replication fork reversal. Our long-term goal is to define the mechanisms that ensure error-free processing of stalled or damaged replication forks, and to understand how to target these mechanisms for the development of new and more effective strategies for cancer treatment. Specifically, our lab made substantial contributions toward the understanding of the mechanism of replication fork reversal that allows replication forks to reverse their course to cope with DNA lesions. We uncovered a key role of the human RECQ1 helicase in the restart of reversed replication forks. We also found that the fork restart function is regulated by poly(ADP-ribose) polymerase (PARP1), which suppresses RECQ1 activity until the damage is repaired, providing the first insight into the molecular steps that drive the timely resolution of reversed replication forks. Shortly thereafter, we identified a second human DNA2- and WRN-dependent mechanism of reversed fork processing and restart. We also found that depletion of the central recombinase RAD51 antagonizes this mechanism, presumably by preventing reversed fork formation. Collectively, our work provided the first mechanistic insight into how replication forks reverse and restart as a pivotal response to treatment with DNA damaging chemotherapeutics, and offers new molecular perspectives to potentiate current DNA-damaging chemotherapeutic regimens by targeting fork reversal.
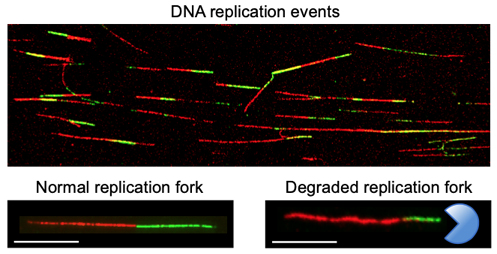
Replication fork protection. Aside from their well-established roles in homologous recombination, the breast cancer susceptibility proteins BRCA1 and BRCA2 are emerging as key factors required for the maintenance of DNA replication fork stability following chemotherapy. We discovered that the main function of BRCA proteins in the context of replication fork stability is to protect reversed replication forks from nuclease-dependent degradation. When BRCA proteins are missing, the reversed DNA forks are unprotected, and nucleases can easily degrade the DNA. This extensive replication fork degradation phenotype has been linked to the sensitivity of BRCA-deficient tumors to agents that damage the DNA or that inhibit specific repair pathways, such PARP inhibitors. To this end, we also discovered that BRCA-deficient cells employ specific fork recovery pathways as a last resort to rescue the degraded forks and withstand DNA-damaging chemotherapy. Following these findings, our next goal is to elucidate the pathways that control fork recovery in BRCA-deficient tumors, and to understand how to target these pathways for the development of new and more effective cancer treatment strategies. Collectively, our studies revisit the functions of central homologous recombination factors in DNA replication and are crucial to understanding how targeting fork recovery pathways can modulate current chemotherapeutic modalities.
Adaptive response to chemotherapeutics. Most of the previous studies, including our own work, investigated how replication is perturbed by DNA-damaging chemotherapeutics following a single- dose treatment, neglecting the fact that patients are treated with multiple doses in a clinical setting. We began to more broadly consider how the replication stress response to DNA damaging chemotherapeutics changes after treatment with multiple drug doses. Using a novel multiple dose approach, we discovered that cells restrain fork reversal when reversed forks cannot be adequately protected by BRCA proteins. In particular, we found that BRCA1-deficient cells adapt to treatment with multiple doses of platinum-based compounds by suppressing fork reversal and promoting PRIMPOL-repriming as an alternative strategy to cope with the drug-induced DNA lesions. This effect is generalizable to other conditions of impaired fork reversal (such as loss of the SMARCAL1 translocase or PARP inhibition) and suggests a new strategy to modulate DNA-damaging chemotherapeutic sensitivity by targeting the PRIMPOL pathway. Our next goal is to expand this analysis to different DNA damaging chemotherapeutics and define whether different adaptive response mechanisms are activated at later time points after drug treatment, depending on the nature of the DNA lesion.
Biosketch
Education
- 1999-1995: Postdoctoral Fellow, Department of Biochemistry and Molecular Biophysics, Washington University School of Medicine, St. Louis, MO
- 1995-1992: PhD in Biochemistry and Molecular Biophysics, University of Padua, Padua, Italy
- 1992-1991: Undergraduate Student, CRIBI Biotechnology Centre, University of Padua, Padua, Italy
- 1992-1987: Laurea in Chemistry, University of Padua, Padua, Italy
- 1990: ERASMUS Exchange Student, Department of Chemistry, University of Newcastle Upon Tyne, UK
Academic Positions & Employment
- present-2019: Adjunct Professor of Biochemistry and Molecular Biology, Saint Louis University School of Medicine, St. Louis, MO
- present-2019: Professor of Medicine and Pathology and Immunology, Section of Medical Oncology, Washington University School of Medicine, St. Louis, MO
- 2019-2014: Professor of Biochemistry and Molecular Biology, Saint Louis University School of Medicine, St. Louis, MO
- 2014-2011: Associate Professor of Biochemistry and Molecular Biology, Saint Louis University School of Medicine, St. Louis, MO
- 2012-2002: Group Leader, International Centre for Genetic Engineering & Biotechnology (ICGEB), Trieste, Italy
- 2002-1999: Staff Scientist, International Centre for Genetic Engineering & Biotechnology (ICGEB), Trieste, Italy
Appointments & Committees
- present-2021: Director, Center for Genome Integrity, Siteman Cancer Center, St. Louis, MO
- present-2015: Member, Siteman Investment Program (SIP) Study Section, Siteman Cancer Center, St. Louis, MO
- present-2015: Co-Leader, DNA Metabolism and Repair (DMR) Group, Solid Tumor Therapeutics Program (STTP), Siteman Cancer Center, St. Louis, MO
- present-2002: Proposal Reviewer, Collaborative Research Program (CRP) – ICGEB Grant
- 2019-2018: Member, Science and Engineering Task Force, Saint Louis University School of Medicine
- 2019-2015: Committee Member, Graduate Program Advisory Council, Graduate Program in Biomedical Sciences, Saint Louis University School of Medicine
- 2019-2017: Permanent Member, Research Planning Committee, Saint Louis University School of Medicine
- 2019-2015: Committee Member, Core Curriculum Committee, Graduate Program in Biomedical Sciences, Saint Louis University School of Medicine
- 2019-2014: Director, Biochemistry Graduate Program, Saint Louis University School of Medicine
- 2015-2014: Director, Doisy Summer Program for Undergraduate Students, Saint Louis University School of Medicine
- 2014-2013: Co-organizer, Biochemistry and Molecular Biology Departmental retreat, Saint Louis University School of Medicine
Honors & Awards
- 2017: Saint Louis University Scholarly Works Award
- 1998-1996: American Heart Association Postdoctoral Fellowship
- 1996-1995: William M. Keck Fellowship, Washington University School of Medicine, St. Louis, MO
Grant Review
- present-2002: ICGEB Research Grant
- 2022: Standing Member, Cancer Etiology Study Section, NCI/NIH
- 2021: Ad Hoc Reviewer, Cancer Etiology Study Section, NCI/NIH
- 2020: Member, NCI Program Project Review Committee
- 2020: Ad Hoc Reviewer, NIA Board of Scientific Counselor’s Meeting
- 2019: Ad Hoc Reviewer, Molecular Genetics A Study Section, NIH
- 2019-2012: President’s Research Fund, Saint Louis University
- 2018-2015: Siteman Cancer Center/Washington University
- 2017: National Institute of Environmental Sciences/NIH, Special Emphasis Panel: Revolutionizing Innovating, Visionary Environmental, Health Research (RIVER)
- 2016: French National Cancer Institute
- 2015: National Science Foundation (NSF), Ad Hoc
- 2015-2011: Wellcome Trust/Cancer Research UK
- 2013: Clinical and Translational Research Funding Program, Barnes-Jewish Hospital Foundation (BJHF)/ Washington University Institute of Clinical and Translational Sciences (ICTS)
- 2013: Medical Research Council UK
- 2003: Human Frontier Science Program
Editorial Boards
- 2018-2015: Member, Editorial Board, Biophysical Chemistry
Professional Societies & Organizations
- present-2009: Member, Italian Society of Biophysics and Molecular Biology
- present-2010: Member, American Society for Microbiology