
Address:
Division of Oncology
Mail Stop 8069-0004-07
Washington University
660 South Euclid Avenue
St. Louis, MO 63110
BJC Institute of Health, Room 7609 (lab)
Ph: 314-747-3898 (lab)
- Tumor suppressors
- Oncogenes
- Ferroptosis
- Breast cancer
- Lung cancer
- Endometrial cancer
1. The collaboration of the ARF and p53 tumor suppressors in preventing triple-negative breast cancer
Triple-negative breast cancer (TNBC) has remained a considerable clinical challenge due to the lack of efficacious genetic targets. We need unique effective therapies and accurate biomarkers that can be used to predict patient responses in TNBC. We find that the ARF tumor suppressor is lost alongside p53 mutation in 60% of TNBC. Potentially stemming from the dual loss of ARF and p53, we have observed that type I IFN signaling is elevated in TNBC. We show that this IFN production is being kept in check by the ADAR1 enzyme. Notably, we discovered that ADAR1 is a novel binding partner for ARF. The central premise of this project is that the novel ARF-ADAR1 interaction provides key insights into how these two proteins function in the etiology of TNBC. The research project focuses on the role of this interaction in regulating the type I interferon response and sensitizing TNBC cells to cell death and immune recognition. The overarching hypothesis of the proposed research is that loss of ARF and p53 results in elevated type I IFN signaling and sensitizes cells to ADAR1 depletion.
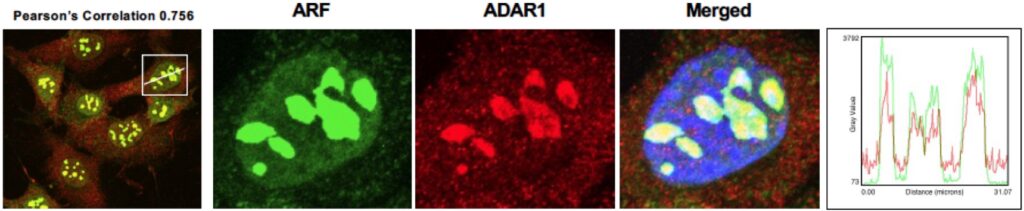
2. Targeting an innate immune signaling pathway in triple-negative breast cancer
Can we identify why some breast cancers become life-threatening metastasis? Can we identify what drives cancer growth and determine how to stop it? These questions have remained at the top of the list for many clinicians, researchers, and patients. Metastasis of cancer cells from the breast to distal target organs is the single-most important cause of mortality in women with breast cancer.
One of the most challenging aspects of eradicating breast cancer and increasing the survival rates of women with breast cancer is the prevention and/or treatment of metastasis. Triple negative breast cancer remains a challenge to clinicians, laboratory investigators, and patients due to its disproportionate number of breast cancer deaths and its lack of an established therapeutic target. Numerous studies have identified potential novel mutational gene targets in TNBC, but single-agent therapeutics have lacked substantial impact in TNBC. More recently, scientists have begun to study ways to activate our own immune system to fight off cancer.
These immune checkpoint inhibitors gained significant clinical traction in breast cancer. Unfortunately, initial promising results have been subsequently overshadowed with failures, particularly in TNBC. In other solid human tumors, the efficacy of immune checkpoint therapies appeared to be enhanced by stimulating our own immune cells into entering the tumor microenvironment in larger numbers to fight the tumor cells. We appear to have found a way to do this effectively in breast cancer by stimulating a pathway controlled by the RIG-I protein. This project will build on our exciting preliminary findings with a series of in vitro and mouse experiments aimed at determining the clinical utility of hyper activating RIG-I and increasing IFN-beta production to sensitize TNBC cells to immune checkpoint therapies.
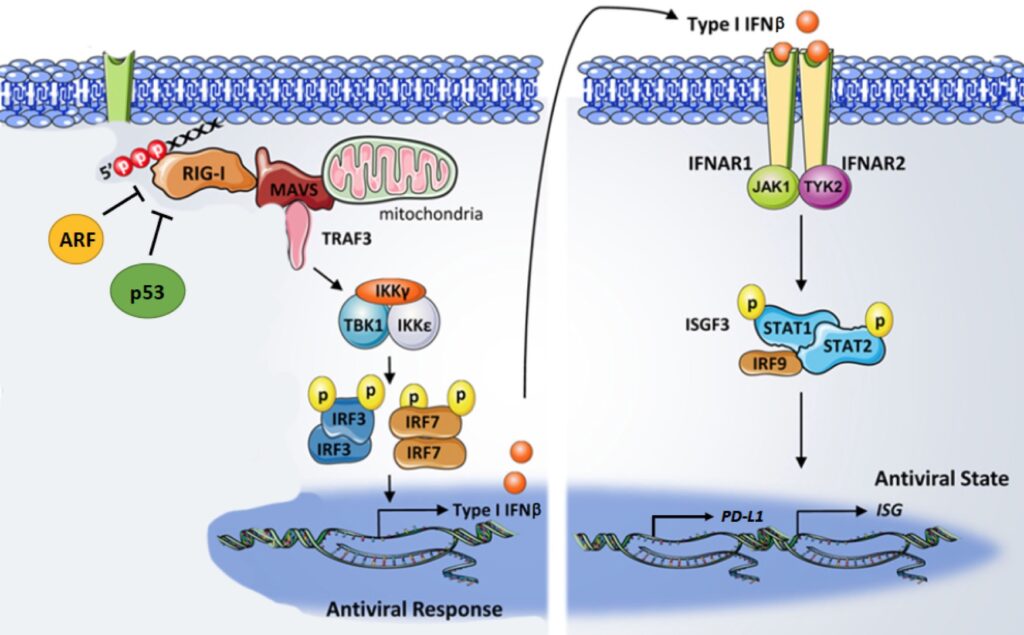
3. Identifying drivers of triple-negative breast cancer in African American women
Triple-negative breast cancer is an overly aggressive breast cancer subtype that disproportionately affects African American (AA) women. AA women have an excessive frequency of metastasis to the lung, liver and brain with generally poor survival. These outcome disparities persist even after controlling for socioeconomic factors such as access to care. Notably, genetic sequencing of breast cancers from women of African and European descent did not find significant somatic mutations that would segregate races. We have identified a novel molecular pathway that appears to preferentially drive TNBC in AA. We found that TNBC from AA women appear to activate an innate immune signaling pathway that culminates in the activation of the ADAR1 oncogene and overexpression of the ISG15 oncoprotein. This fundamental finding could point to a novel series of treatment targets for AA TNBC. The lab is now poised to study how ISG15 conjugation to proteins alters disease progression in AA women.
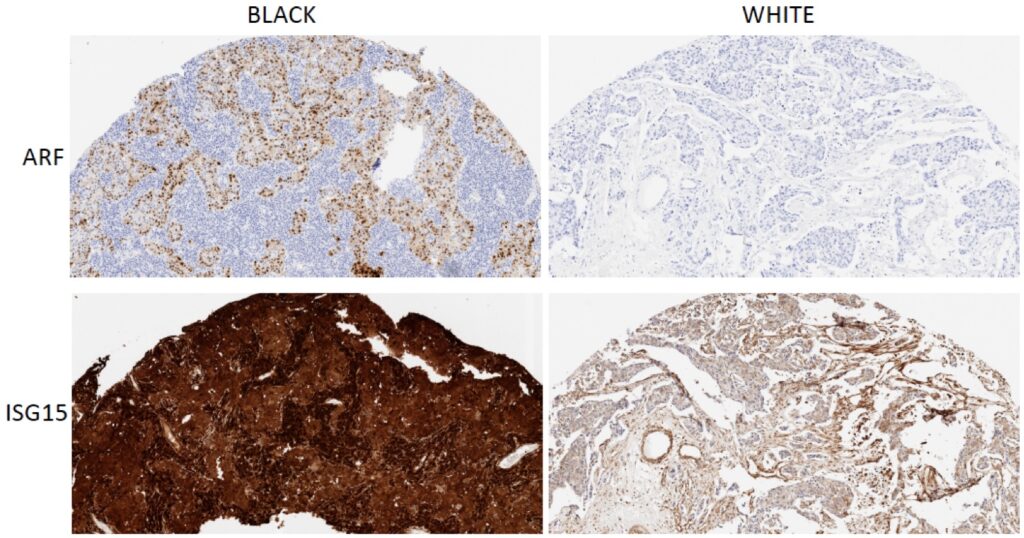
4. Using high-throughput DNA-barcoded chemical libraries to screen for oncogene inhibitors
The RNA editing enzyme ADAR is a valuable therapeutic target for multiple cancers, including triple-negative breast cancer. ADAR-dependent cancer cell lines share a signature of elevated interferon stimulated gene expression, potentially making it possible to classify ADAR dependent tumors. However, there are currently no selective inhibitors of ADAR. The goal of this project is to identify small molecule inhibitors of ADAR. These ADAR inhibitors will be evaluated for their ability to inhibit ADAR and arrest growth of TNBC cell lines in vitro and tumorigenesis of patient-derived xenograft models of TNBC in vivo. Eventually this work could lead to a clinical trial of ADAR inhibitors for the treatment of TNBC, with patients further stratified by tumoral interferon stimulated gene expression to identify likely responders. Beyond the cell intrinsic effects of ADAR depletion in ADAR-dependent cell lines, it has been shown that depletion of ADAR can overcome resistance to immune checkpoint blockade. This observation supports combining immune checkpoint inhibitors targeting PD-1 or PD-L1 with ADAR inhibitors. Inhibition of ADAR could be the first broadly applicable targeted therapy for the treatment of TNBC. Through this project we will identify and validate a small molecule inhibitor of ADAR, paving the way for clinical trials of ADAR inhibitors for the treatment of TNB.
5. Design and fabrication of novel microfluidic devices to model lymphatics in breast cancer metastasis
Regional lymph nodes are the primary sites of lymphatic drainage from all areas of the breast, and the extent of their involvement in breast cancer is a strong predictor of disease relapse and patient survival. Recent studies demonstrate that lymphatic vessels undergo dynamic remodeling when co-cultured with tumor cells, which facilitates metastasis. Lymphatic vessel contribution has not yet been evaluated for any racial bias in TNBC. Although much has been learned about the distinct mutational landscape of TNBC tumor cells, the underlying molecular and cell biological mechanisms that drive the apparent racial disparity remain unknown. Currently, a major obstacle in the field is that accurate models of racially diverse TNBC and its lymphatic microenvironment do not exist, underscoring the overall difficulty in identifying drivers of aggressive African American disease. In this project, we will develop a series of innovative TNBC-on-a-chip microfluidic devices in which to study potential racial drivers of TNBC phenotypes using patient-derived tumor xenografts and patient-derived lymphatic endothelial cells.
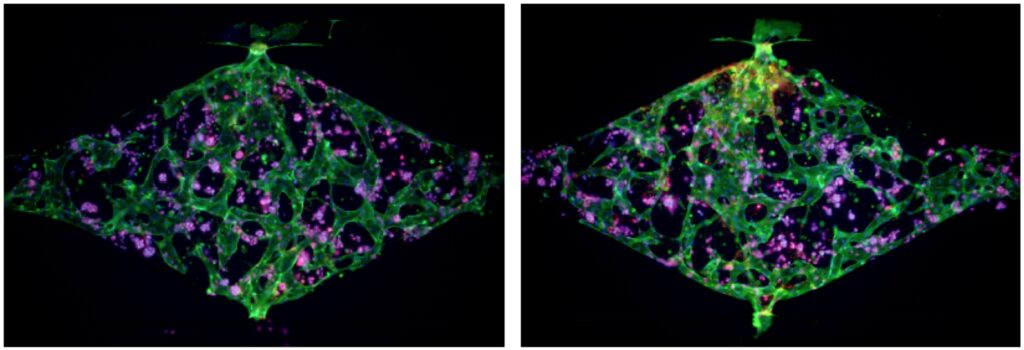
6. Functional interaction of ADAR1 and MDM2 oncogenes in ferroptosis
The biggest challenges in breast cancer therapy include the lack of targeted therapies for triple-negative breast cancer, and endocrine resistance developed during the treatment of hormone-responsive breast cancer. TNBC accounts for 15-20% of all breast cancer cases and is more aggressive with worse prognosis due to the lack of options for targeted therapies. Conversely, 80% of breast cancers express hormone receptors (ER and/or HER2) and can be treated with endocrine or HER2-targeted therapies with considerable success, especially for early-stage cancers. However, acquired endocrine resistance remains a major issue that occurs in >30% of patients receiving hormone-based therapy, often followed by distant cancer metastasis. Therefore, development of more specific and actionable targets remains a top priority.
With the ability to change DNA-encoded genetic information after transcription, site-specific substitution of RNA, or RNA editing, could result in significant physiological consequences. As the most well-studied RNA editing enzyme, adenosine deaminase acting on RNA 1 (ADAR1) can drive transcriptomic and proteomic diversity through A(adenosine)-to-I(inosine) RNA editing in human diseases, including cancer. We recently demonstrated an elevated ADAR1-dependent phenotype shared by TNBC, highlighting its potential as a novel therapeutic target. Through RNA sequencing, we identified significant MDM2 upregulation among all cell lines (TNBC and non-TNBC) upon ADAR1 loss. It has recently been shown that MDM2 plays paradoxical roles in cancer, acting as either a therapeutic target with pro-tumorigenic activities, or a tumor-suppressive factor by promoting lipid remodeling and iron-dependent cell death (ferroptosis). We hypothesize that ADAR1 loss in breast can sensitize cancer cells to 1) ferroptosis through induced lipid remodeling, or 2) MDM2 inhibitory treatments through MDM2 upregulation, dictated by their subtypes.
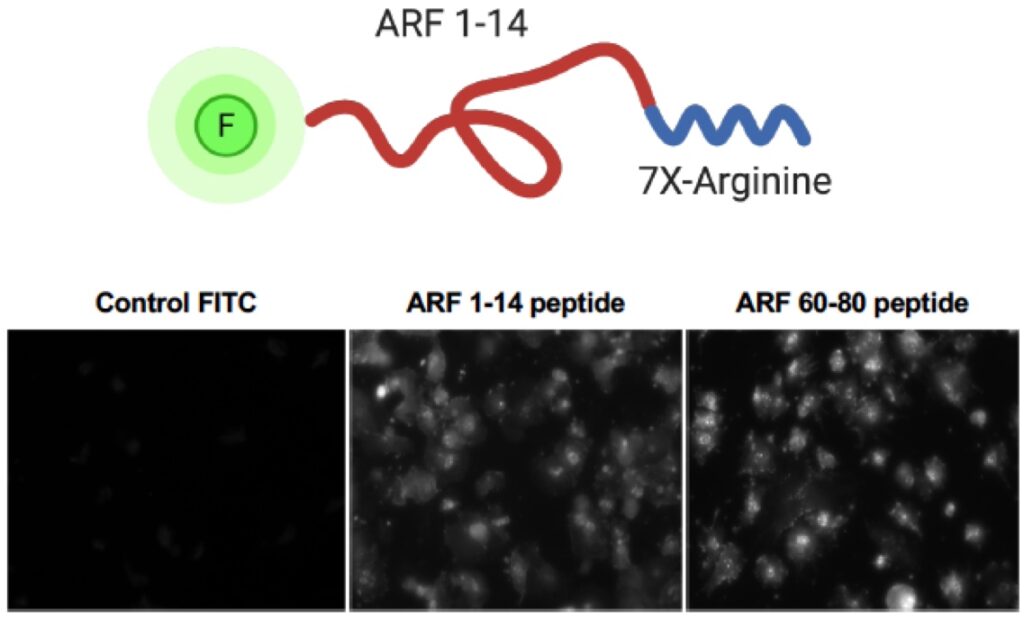
7. Restoration of ARF tumor suppressor function through peptide mimics
Cancer biologists have long sought to use genetic alterations of tumor cells as novel therapeutic targets. Nearly every anti-cancer drug that has been brought to the clinic using this ideal has targeted oncogenes whose altered activity is thought to drive cancer formation and progression. However, these agents have not appreciably altered the 5-year survival rate of lung cancer. We now seek to switch this thinking. For current and future generations, the prevention or treatment of lung cancer represents a tangible goal in the eradication of this deadly disease. While this goal seems attainable on the surface, the complexity of lung cancer on both a genomic and physiologic level tends to set our sights on the very distant future in reaching this goal. However, recent advances in whole genome sequencing and molecular techniques has provided invaluable tools in the dissection of the lung cancer process. Moreover, the identification of key oncogene and tumor suppressor networks in lung cancer has opened strategic windows to improved patient care. We now propose a highly innovative notion that restoring the ARF tumor suppressor, the second-most mutated tumor suppressor in non-small cell lung cancer (NSCLC), can be clinically impactful. We propose numerous innovative tools and experimental approaches aimed at opening the tumor suppressor field to clinical lung cancer treatment avenues.
8. Collaborative regulation of translational targets by the ARF and NF1 tumor suppressors
Plexiform neurofibromas (PNs) arising from Schwann cells are a hallmark manifestation of NF1. Approximately 15% of PNs will transform into very aggressive late-stage malignant peripheral nerve sheath tumors (MPNSTs) which are a leading mortality in NF1 patients. A gene was mapped to individuals affected by NF1. This gene, termed Nf1, carried an inheritable mutation that appeared to predispose these individuals to NF1. Because of the association between NF1 and tumor development, Nf1 has been classified as a tumor suppressor gene. The product of the Nf1 gene is the neurofibromin protein. Work has shown that the normal role of neurofibromin is to prevent the hyperactivation of the mTOR growth signaling pathway. Loss of neurofibromin in NF1 individuals leads to uncontrolled mTOR activities. This in turn directs Schwann cells to initially grow (get larger) and proliferate (divide). However, Nf1-deficient cells rapidly induce the ARF tumor suppressor which prevents further growth and proliferation. In this manner neurofibromin and ARF work together to prevent Schwann cell transformation and progression to MPNST.
While much of the NF1 scientific community is focused on the mTOR pathway as a sole manifestation of NF1, our group has chosen to study the mechanisms underlying the progression from PN to MPNST through the concomitant loss of both the Nf1 and Arf tumor suppressors. We have shown that this progression involves an increase in mRNA translation. Once bound to an mRNA, the ribosome translates the message into protein. One could imagine that if mRNAs are loaded onto ribosomes faster, then more protein will be produced. Likewise, an mRNA that is loaded more slowly, or not at all, would result in less protein synthesis. In this manner, regulation of which mRNAs are being translated becomes integral in determining the protein landscape of the cell, and ultimately what processes the cell performs. The goal of this project is to identify the mRNAs whose translation is affected by the loss of Nf1 and Arf, and to characterize the mechanism behind their altered translation. To achieve this goal, we are using two complimentary approaches. The first involves a novel screen, one that has never been performed in the Neurofibromatosis field, although our lab has already utilized this technique in other cell types. Our screen will serve to identify mRNAs that are either loaded faster, slower or not at all on ribosomes in Nf1/Arf-deficient Schwann cells. The second approach involves understanding how these mRNAs are loaded differently by identifying proteins that uniquely interact with these mRNA. We hypothesize that specific proteins are responsible for binding to these mRNAs and either placing them on the ribosome or preventing them from loading onto the ribosome. If we can identify these translation regulators, then we may discover why PNs progress to MPNST following loss of Nf1 and Arf tumor suppressor genes.
Biosketch
Education
- 1997-1994: PhD in Cell & Molecular Biology, Saint Louis University, St. Louis, MO
- 1993-1989: BS in Biotechnology, Bradley University, Peoria, IL
Academic Positions & Employment
- present-2022: Professor, Biology, Washington University, St. Louis, MO
- present-2017: Professor, Departments of Medicine and Cell Biology & Physiology, Washington University, St. Louis, MO
- 2017-2007: Associate Professor, Departments of Medicine and Cell Biology & Physiology, Washington University, St. Louis, MO
- 2007-2001: Assistant Professor, Departments of Medicine and Cell Biology & Physiology, Washington University, St. Louis, MO
- 2001-1997: Associate, Howard Hughes Medical Institute, Tumor Cell Biology Lab, St. Jude Children’s Research Hospital (PI, Charles J. Sherr)
- 1994-1993: Scientist, Monsanto/G.D. Searle, Immunoinflammatory Diseases Research Group
University Committees
- present-2019: Co-Director, Cancer Biology Graduate Program, Washington University
- present-2012: Internal Rita Allen Scholars Selection Committee, Washington University
- present-2012: Internal Pew and Searle Scholars Selection Committee, Washington University
- present-2011: Chair ACS-IRG Committee, Washington University
- present-2010: Founding Member, BRIGHT/ICCE Institute, Washington University
- present-2010: Co-Leader, Breast Cancer Research Program, Siteman Cancer Center, Washington University
- present-2006: American Cancer Society-IRG Committee, Washington University
- present-2006: Founding Member, Breast Cancer Research Program, Siteman Cancer Center
- present-2004: Division of Biological and Biomedical Sciences Admissions Committee
- 2019-2009: Co-Director, Molecular & Cellular Biology Graduate Program, Washington University
- 2019-2006: Division of Biological and Biomedical Sciences, Molecular Cellular Biology Steering Committee
- 2015-2012: Tumor Tissue Bank Committee, Washington University
- 2015-2009: Molecular Imaging Center, Grant Review Committee, Washington University
- 2014-2002: Neuro-Oncology Research Group, Founding Member
- 2013-2001: Cellular Proliferation Program, Siteman Cancer Center
- 2012: Internal Ralph E. Powe Award Selection Committee, Washington University
- 2010-2005: Siteman Cancer Center Urology Research Group
- 2009-2006: Division of Biological and Biomedical Sciences, Assistant Director for Recruiting
- 2009-2006: Division of Biological and Biomedical Sciences Admissions Committee, Co-Chair
Honors & Awards
- present-2023: Department of Defense, Breast Cancer Research Program, Member of Programmatic Panel
- 2016: American Cancer Society, Honorary Corporate Hero
- 2016-2015: Department of Defense, Breast Cancer Research Program, Invited Integration Panel Member
- 2016-2014: The Artemis 2020 Project, Invited Member
- 2014, 2013, 2012, 2011, 2010, 2009, 2008, 2007, 2006, 2005 Distinguished Service Teaching Award, First Year Medical Students, Washington University
- 2008: American Cancer Society Scholar
- 2008: CDMRP, Era of Hope Scholar in Breast Cancer Research
- 2007: Teacher of the Month, Medical Student Class of 2009
- 2002: Pew Scholar in Biomedical Sciences
- 2001: Edward Mallinckrodt, Jr. Foundation Scholar
- 1993: Monsanto/G.D. Searle Competitive Summer Internship
- 1993: Bradley University Outstanding Research Award
National Committees (AD Hoc Member)
- present-2012: National Institutes of Health/NCI, ZRG Special Emphasis Panel on Oncological Sciences
- 2020: National Institutes of Health/NCI Tumor Cell Biology Study Section
- 2019: National Institutes of Health/NCI ZRG1-OBT R21 Study Section, Committee Assistant Chair
- 2019-2018: National Institutes of Health/NCI, ZRG1 OBT R15 Study Section
- 2017: National Institutes of Health/NCI, ZRG1 CSBC U54 Section
- 2016: National Institutes of Health/NCI, ZRG1 OBT Study Section
- 2015: National Institutes of Health, Graduate Program Training Grant, UT Southwestern, Reviewer
- 2014: Health Research Council of New Zealand, Investigator Grants, International Reviewer
- 2011: Austrian Science Fund, Investigator Grants, International Reviewer
- 2011, 2010, 2007: Cancer Research UK Investigator Grants, International Member
- 2010: National Medical Research Council, Singapore, Investigator Grants, International Reviewer
- 2009: American Association for Cancer Research Annual National Meeting, Organizer and Reviewer for Cell Growth Pathways
- 2009: National Institutes of Health/NCI, TCB Study Section
- 2009-2008: National Institutes of Health, ZRG Special Emphasis Panel
- 2007: National Institutes of Health/NCI, MONC Study Section
- 2006: National Institutes of Health, Cell Signaling and Dynamics Study Section
National Committees (Permanent Member)
- present-2022: National Institutes of Health/NCI, ZRG-F09A F30/31/32 Fellowships: Oncology Study Section, Chair
- present-2019: Medical University of South Carolina Hollings Cancer Center Grants, External Reviewer
- present-2015: American Cancer Society, Heartland Association, Research Liaison
- present-2015: Department of Defense-CDMRP, Breast Cancer Research Program, Integration Panel Member
- present-2012: The Ohio State University Cancer Center, Investigator Grants, External Reviewer
- 2021-2019: National Institutes of Health/NCI, ZRG-F09A F30/31/32 Fellowships: Oncology Study Section, Assistant Chair
- 2018-2014: The Artemis 2020 Project, Member
- 2015-2013: Komen for the Cure, Postdoctoral Research Grants, Reviewer
- 2014-2012: Department of Defense-CDMRP, Neurofibromatosis, Molecular Biology Study Section, Reviewer
- 2008-2005: Department of Defense, Army, SBIR Proteomics and Neoplasias Committee
- 2005-2002: American Heart Association, Cell Transport Study Section
Professional Societies
- American Society for Microbiology
- American Association for Cancer Research
- American Cancer Society